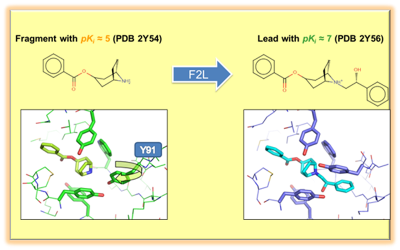
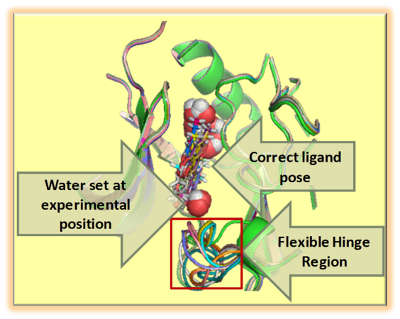
Description
An evolutionary tuning of optimal operational parameters for the libsvm Support Vector Machine tool.
Libsvm-GAconfig is a Unix script-driven package for the evolutionary search of optimal libsvm operational parameters, leading to Support Vector Machine models of maximal predictive power and robustness. Unlike common libsvm parameterizing engines, the current distributions includes the key choice of best-suited sets of attributes/descriptors, next to the classical libsvm operational parameters (kernel choice, cost, etc.), allowing a unified search in an enlarged problem space. It relies on an aggressive, repeated cross-validation scheme to ensure a rigorous assessment of model quality. This package also allows the search for optimal ISIDA descriptors and optimal hyper-parameters for the Generative Topographic Mapping approach, two methods widely developed in the laboratory of Chemoinformatics. The approach is versatile, covering both regression and classification problems and supporting a large variety of parallel deployment schemes. It was, for example, successfully used to generate very large (> 9000 instances) dataset-based chemogenomics models.
Screenshots of results
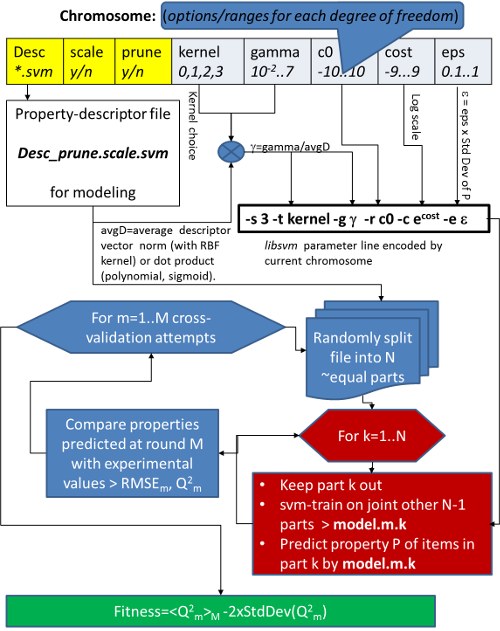
Platforms
Linux64 command line
Documentation
Documentation is available here
Authors
D. Horvath, J.B. Brown, G. Marcou, A. Varnek
Bibliography
D. Horvath, J.B. Brown, G. Marcou and A. Varnek
An Evolutionary Optimizer of libsvm Models
Challenges, 2014, 5(2), 450-472
Download
Please please fill the form available here to request a download link for this software.
However, since libsvm-GAconfig is under steady development, it is advised to contact Dragos Horvath (dhorvath@unistra.fr) for the latest versions.